Sequence comparison helps comparing two or more things in biological data and allows studying how closely related they might be, may be in terms of function, evolution, or both.
Most commonly used type of comparison in bioinformatics is ‘sequence comparison’. It works work out how closely a nucleotide or protein sequence is related is to others in the public databases. This can be performed by aligning the sequences, rearranging them in order to find the best match possible, and takes into note the insertions, deletions, and substitutions that may have developed since divergence from a theoretical common ancestor.
Dot plot method
Dot plot, one of the simplest ways of comparing two sequences, also called as a strip plot or dot chart. Put one of the sequences horizontal and the other as vertical. Make a dot (or some other point mark) in the space between, wherever the sequences have the same symbol such as nucleotide or amino acid. This is very simple and fast, and gives a good visual overview and is also useful for example when comparing genomes. In the dot plot for two bacterial genomes L. acidophilus and L. bulgaricus the disadvantage can include that it may not be able to identify the best possible alignment.
Sequence alignments
Alignment is a way of arranging DNA, RNA and protein sequences so that similar regions are highlighted. They are used for finding conserved and important positions, since these will be visible in the alignment. They can also be used when doing homolog search in nucleotide or amino acid sequence databases. Unlike dot plots, the algorithms for alignment use different ways of modeling evolution.
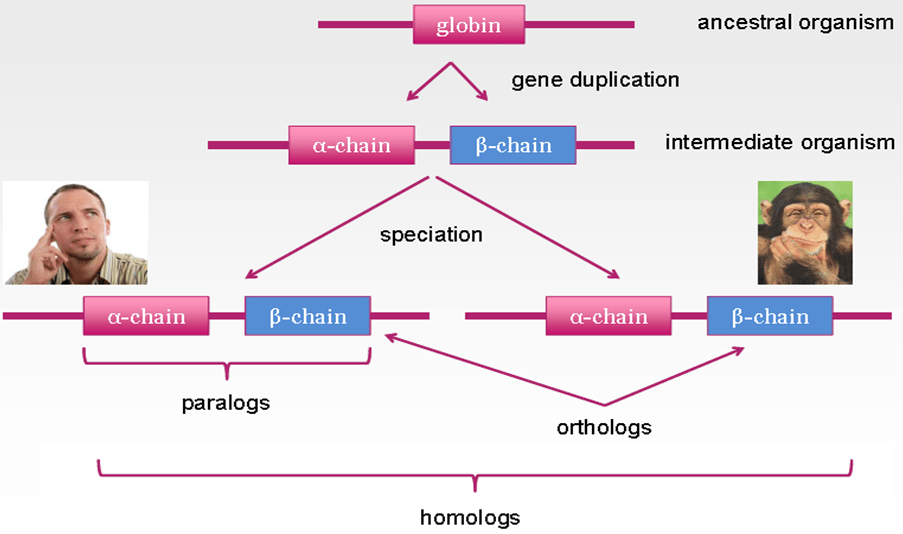
Homologs, paralogs and orthologs
When using bioinformatics tools to understand evolution (such as alignments of phylogenetic trees) it is important to keep in mind that genes may be related to each other in different ways. Homologs are genes/proteins that have evolved from the same ancestral gene/protein whereas; paralogs and orthologs are the two different kinds of homologs. Paralogs appear from gene duplication. For example, the human alpha-chain and human beta-chain hemoglobins are paralogs as they are located in the same species.
Orthologs appear from speciation events. For example, the human alpha-chain hemoglobin and the chimpanzee alpha-chain hemoglobin are orthologs. They are present in different species, but originated in a common ancestor species. When comparing the sequences of the genes, see the evolutionary history being reflected in their levels of similarity. For example, chimpanzee and the human alpha chain hemoglobin’s have almost identical amino acid sequences, even though they take place in the two different species. The human alpha and beta hemoglobin sequences, on the other hand, differ quite a lot, although they are found in the same genome. This pattern also points that the human chimp speciation event occurred quite recently, whereas the event of alpha beta duplication took place a much longer time ago. This example illustrates the usefulness of sequence alignment.
Types of alignments
Alignments can be classified according to global vs. local, pairwise vs. multiple, nucleotide vs. protein, and exact vs. heuristic. Additional categories are possible.
In ungapped alignments, only complete sequences can be shifted to the left right to compute scores while in gapped alignments, gap symbols can be added to represent two types of point mutation, namely insert and delete. In the example, the ungapped alignment gets a score of 4 because there are 4 positions where the two sequences have identical symbols (A vs A in the first two positions, A vs A in the sixth position, and T vs T in the fifth position). The two examples of gapped alignments show the many more possibilities and that moving the gaps a few positions can have a large impact on the alignment score.

A simple scoring scheme takes only identity into account, setting for example +1 for match, 0 for mismatch and -1 for gap symbols. This simple scoring only works for nucleotide sequences.
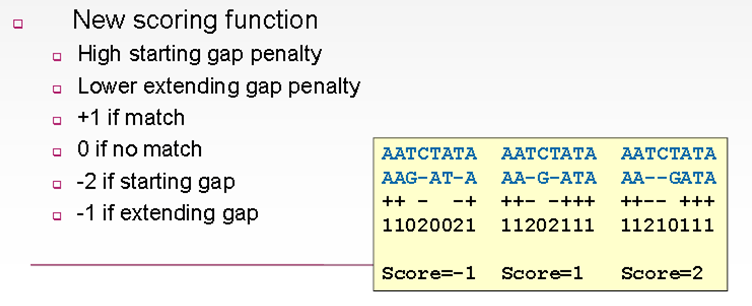
A large number of the alignment programs permit the user to set two sorts of gap penalty, first for opening a new gap and second for extending an already existing gap. How does this reflect evolution? Mutations that are involved in the insertion or deletion of a stretch of amino acids occur frequently. In case the penalty for a gap of only one nucleotide is 1, then having the penalty for a gap of three nucleotides being three times greater (i.e. 3) is not accurate. The alignments are meant to illustrate that a single gap of length two is preferred over two separate gaps, each of length one. The third (rightmost) alignment gives a slightly higher overall score (i.e. 2) than the other two alignments. An alignment tool might have a few gap opening penalty such as 15 and gap extending penalty 2. When using LALIGN in projects, it is better to pay special attention to the default values of these two penalties, and alterations to the other values if needed.
Examples of global pairwise protein alignments
The orthologs HBA_HUMAN and HBA_PANTR are globally aligned. It is found that they are 100% identical. So the human and chimpanzee alpha-chain hemoglobins are exactly equal. The paralogs HBA_HUMAN and HBB_HUMAN are then aligned. They can be found to be identical at 43.6% of the positions in the alignment. Matches between identical amino acids are pointed with the help of two dots, matches between similar amino acids are pointed by one dot, and matches between non-similar amino acids (i.e. mismatches) are not pointed by any dots. The gap symbols have to be added in a few places.
Examples of local and global alignments
The human and frog lipocalin proteins can be aligned both globally and locally. In the local alignment, the identity increased slightly, from 26.7% to 27.6%. The local alignment identifies a conserved sub region which might not be visible in the global alignment. It begins with TADG, showing from the position at 96 in the human sequence, and from the position at 66 in the frog sequence. In general, local alignments can identify conserved regions that are missed by global alignment.
Dynamic programming
The standard pairwise alignment algorithms, Needleman-Wunsch (global) and SmithWaterman (local), both use the same strategy. Dynamic programming includes breaking down a large problem into smaller sub-problems. This strategy guarantees finding the highest scoring alignment, but has the disadvantage of being too slow for multiple alignments.